Artículo
Revelando la brecha de evidencia: Medicamentos huérfanos vs. medicamentos no huérfanos en las evaluaciones de tecnologías sanitarias alemanas
-
Kristina Dittrich, PhD
-
Nadine Klusmeier, PhD
-
Mareike Kubinski, PhD
-

Jan-Frederik Löpmeier-Röh, MSc
-

Susanne Kossow, PhD
-
Michael Reinartz, PhD
-

Werner Kulp, PhD
A menudo se prejuzga que los medicamentos para enfermedades raras carecen de evidencia porque tienen un beneficio adicional garantizado por ley. Pero, ¿está justificado este prejuicio? Este artículo lo lleva a una exploración esclarecedora de la evaluación de tecnologías sanitarias en Alemania para dilucidar la realidad detrás de estas suposiciones persistentes.

Enfermedades raras y sus tratamientos en la ETS alemana
Las enfermedades raras, que por definición afectan solo a 1 de cada 2.000 personas en la Unión Europea, a menudo carecen de opciones de tratamiento eficaces. Los medicamentos huérfanos (DO) llenan este vacío y, por lo tanto, reciben incentivos especiales para su desarrollo y comercialización. Para el acceso a los mercados en Alemania, las DO reciben un tratamiento único en la evaluación de tecnologías sanitarias (ETS).
La HTA alemana para medicamentos está regulada por la «Arzneimittelmarktneuordnungsgesetz» (Ley de Reorganización del Mercado Farmacéutico, AMNOG). AMNOG tiene como objetivo equilibrar el reembolso para los fabricantes farmacéuticos y la carga de gasto para el sistema nacional de salud. Los medicamentos están obligados a someterse a una evaluación de los beneficios en comparación con una terapia estándar existente (la llamada terapia [ACT]comparativa adecuada). La evaluación de los beneficios es obligatoria cuando se lanza un medicamento recién autorizado con un nuevo principio activo en Alemania o para las indicaciones recientemente autorizadas de un medicamento ya evaluado. Sobre la base de la evidencia clínica, el Comité Federal Conjunto (G-BA, por sus siglas en inglés) evalúa el beneficio médico adicional utilizando seis categorías ('mayor', 'considerable', 'menor', 'no cuantificable', 'sin beneficio adicional' o 'menor').
En el caso de los OD, la ley otorga un beneficio médico adicional, y la evaluación de la medida se basa en pruebas fundamentales sin la obligación de compararlas con un ACT. Sin embargo, si las ventas alcanzan los 30 millones de euros o la indicación pierde la condición de huérfana, la reevaluación según el proceso de evaluación estándar pasa a ser obligatoria. Los medicamentos no huérfanos (no OD), que están obligados a la evaluación periódica de los beneficios, también pueden ser objeto de una nueva evaluación, ya sea de forma obligatoria en caso de decisiones temporales (por ejemplo, la G-BA solicita datos adicionales) o a petición del fabricante farmacéutico.
La evaluación de las prestaciones es la base para la negociación del precio del reembolso entre el fabricante farmacéutico y la Asociación Nacional de Fondos Obligatorios del Seguro de Enfermedad (GKV-SV). Solo los medicamentos con un beneficio médico adicional pueden alcanzar un precio más alto que los costos del ACT en la negociación de precios. Dentro de los primeros seis meses después del lanzamiento, el fabricante es libre de definir el precio de lanzamiento, mientras que el precio negociado se aplica a partir del séptimo mes. El precio de reembolso de un medicamento puede cambiar aún más con el tiempo cuando las reevaluaciones y las nuevas indicaciones para el medicamento respectivo pasan por HTA y las negociaciones de precios posteriores.
Este artículo explora los resultados de un análisis en profundidad de las ETS alemanas centrado en la comparación de los criterios de evidencia y los precios entre las DO y las no DO (tanto para la evaluación inicial como para la reevaluación) y tiene como objetivo arrojar luz sobre si las DO carecen de evidencia en la ETS en comparación con las que no lo son.
En una revisión exhaustiva se analizaron las evaluaciones de los beneficios para los DO y los no DO desde 2011 hasta mayo de 2024 a partir de una base de datos de G-BA. Este análisis incluyó 959 evaluaciones de beneficios. De estos, 810 fueron evaluaciones iniciales de medicamentos recientemente autorizados o indicaciones recientemente autorizadas de un medicamento, mientras que 149 fueron reevaluaciones, y algunos productos se reevaluaron más de una vez para una indicación respectiva. El análisis de 810 evaluaciones iniciales de beneficios reveló que el 27% de las evaluaciones evaluaron las DO, mientras que el 73% no fueron DO. En total, se realizó una nueva evaluación para el 15% de todas las evaluaciones (7% de DO y 8% sin DO).
Todas las reevaluaciones se compararon con la evaluación inicial de beneficios respectiva, si correspondía, para alinear las evaluaciones relacionadas con el mismo medicamento. Los datos se extrajeron para los DO y los no DO, centrándose en parámetros como la evidencia evaluada (tipos de estudios, número de estudios y tamaño de los estudios), el nivel de beneficio adicional otorgado y el precio. Las evaluaciones se centraron en los casos en los que se realizó una evaluación inicial (60 DO y 64 no OD) y una reevaluación (54 DO y 70 no OD, lo que reflejó que algunos fármacos perdieron el estado de DO desde el punto de vista de la prevalencia y se reevaluaron con arreglo al procedimiento estándar como no OD) para determinar si las pruebas, el nivel de beneficio y el precio cambiaron entre la evaluación inicial y la reevaluación. y comparar entre los DO y los que no lo son.
En el caso de los OD, la ley otorga un beneficio médico adicional, y la evaluación de la medida se basa en pruebas fundamentales sin la obligación de compararlas con un ACT. Sin embargo, si las ventas alcanzan los 30 millones de euros o la indicación pierde la condición de huérfana, la reevaluación según el proceso de evaluación estándar pasa a ser obligatoria. Los medicamentos no huérfanos (no OD), que están obligados a la evaluación periódica de los beneficios, también pueden ser objeto de una nueva evaluación, ya sea de forma obligatoria en caso de decisiones temporales (por ejemplo, la G-BA solicita datos adicionales) o a petición del fabricante farmacéutico.
La evaluación de las prestaciones es la base para la negociación del precio del reembolso entre el fabricante farmacéutico y la Asociación Nacional de Fondos Obligatorios del Seguro de Enfermedad (GKV-SV). Solo los medicamentos con un beneficio médico adicional pueden alcanzar un precio más alto que los costos del ACT en la negociación de precios. Dentro de los primeros seis meses después del lanzamiento, el fabricante es libre de definir el precio de lanzamiento, mientras que el precio negociado se aplica a partir del séptimo mes. El precio de reembolso de un medicamento puede cambiar aún más con el tiempo cuando las reevaluaciones y las nuevas indicaciones para el medicamento respectivo pasan por HTA y las negociaciones de precios posteriores.
Este artículo explora los resultados de un análisis en profundidad de las ETS alemanas centrado en la comparación de los criterios de evidencia y los precios entre las DO y las no DO (tanto para la evaluación inicial como para la reevaluación) y tiene como objetivo arrojar luz sobre si las DO carecen de evidencia en la ETS en comparación con las que no lo son.
En una revisión exhaustiva se analizaron las evaluaciones de los beneficios para los DO y los no DO desde 2011 hasta mayo de 2024 a partir de una base de datos de G-BA. Este análisis incluyó 959 evaluaciones de beneficios. De estos, 810 fueron evaluaciones iniciales de medicamentos recientemente autorizados o indicaciones recientemente autorizadas de un medicamento, mientras que 149 fueron reevaluaciones, y algunos productos se reevaluaron más de una vez para una indicación respectiva. El análisis de 810 evaluaciones iniciales de beneficios reveló que el 27% de las evaluaciones evaluaron las DO, mientras que el 73% no fueron DO. En total, se realizó una nueva evaluación para el 15% de todas las evaluaciones (7% de DO y 8% sin DO).
Todas las reevaluaciones se compararon con la evaluación inicial de beneficios respectiva, si correspondía, para alinear las evaluaciones relacionadas con el mismo medicamento. Los datos se extrajeron para los DO y los no DO, centrándose en parámetros como la evidencia evaluada (tipos de estudios, número de estudios y tamaño de los estudios), el nivel de beneficio adicional otorgado y el precio. Las evaluaciones se centraron en los casos en los que se realizó una evaluación inicial (60 DO y 64 no OD) y una reevaluación (54 DO y 70 no OD, lo que reflejó que algunos fármacos perdieron el estado de DO desde el punto de vista de la prevalencia y se reevaluaron con arreglo al procedimiento estándar como no OD) para determinar si las pruebas, el nivel de beneficio y el precio cambiaron entre la evaluación inicial y la reevaluación. y comparar entre los DO y los que no lo son.

Comparabilidad de las pruebas presentadas para los DO y los que no lo son
En el caso de los OD, la ley otorga automáticamente un beneficio adicional. Por lo tanto, ¿qué evidencia clínica se presenta para los DO en las evaluaciones de los beneficios y en qué se diferencia de las evaluaciones sin DO? Como primer paso en las evaluaciones estándar de beneficios, el G-BA evalúa la calidad de los estudios presentados en función del nivel de evidencia para proporcionar un nivel de conocimiento suficientemente confiable. Sobre la base de esta evaluación, el estudio se acepta o no se considera en la evaluación. Además, los estudios previstos para una evaluación estándar de los beneficios deben incluir una comparación del medicamento con el TCA, definido por la G-BA. Mientras que las DO se benefician de un criterio menos estricto de ejecución de las pruebas para las evaluaciones iniciales, las que superan el límite de ventas se vuelven a evaluar con arreglo al procedimiento estándar, incluso si conservan la condición de huérfanas desde el punto de vista de la prevalencia.
Las evaluaciones mostraron que los ensayos controlados aleatorios (ECA), el estándar de oro de la evidencia, fueron el tipo de estudio predominante tanto para los DO como para los que no lo fueron (Figura 1). En el caso de los no DO, el G-BA aceptó más ECA en la reevaluación (51/70 casos, 73%) que en la evaluación inicial (44/64 casos, 69%), mientras que en el caso de los DO fue a la inversa (45/60 evaluaciones iniciales, 75%; 35/54 reevaluaciones, 65%).
Inicialmente, los estudios clínicos presentados por el fabricante fueron aceptados por el G-BA en todas las evaluaciones de OD; la mayoría de los estudios (45/60 casos, 75%) fueron ECA y el 25% (15/60 casos, 25%) no fueron ECA (p.ej. ensayos de un solo grupo, tamaños de estudio pequeños, uso de resultados indirectos). Cuando los DO se reevaluaron en una evaluación estándar de beneficios, habiendo excedido el límite de ventas, la proporción de ECA aceptados y no ECA disminuyó al 65% (35/54 reevaluaciones) y al 13% (7/54 reevaluaciones), respectivamente. En 12 de las reevaluaciones de DO (22%), los estudios respectivos no fueron aceptados por el G-BA, lo que refleja los criterios de evidencia más estrictos aplicados en el proceso de evaluación estándar. El incumplimiento del ACT podría ser la razón principal de este cambio.
A diferencia de los DO, se aceptaron muy pocos ECA en las evaluaciones de los beneficios no relacionados con el DO (2/64 evaluaciones iniciales; 1/70 reevaluaciones). Por lo tanto, parece más probable que se acepten los ECA para los DO que para los que no lo son. Además, la evidencia del fabricante fue aceptada en todas las evaluaciones iniciales de DO, mientras que los estudios presentados fueron rechazados en 18 evaluaciones y reevaluaciones de no GO. Esto se debe al hecho de que en las evaluaciones de DO los estudios fundamentales siempre son aceptados por la ley.
Figura 1. Distribución del tipo de estudio entre DO y no DO en la evaluación inicial (AI) y la reevaluación (AR)
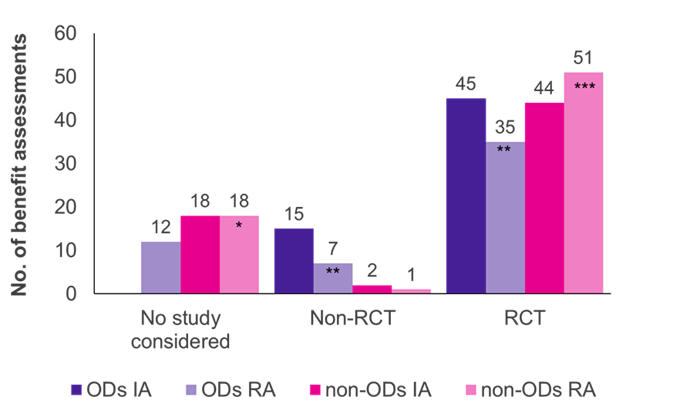
Llave: IA – evaluación inicial; No. –número; OD – medicamento huérfano; RA – reevaluación; ECA: ensayo controlado aleatorizado.
Además, y como era de esperar, hubo una correlación positiva entre un mayor tamaño de las poblaciones de estudio y la disponibilidad de un ECA (datos no mostrados).
Inicialmente, los estudios clínicos presentados por el fabricante fueron aceptados por el G-BA en todas las evaluaciones de OD; la mayoría de los estudios (45/60 casos, 75%) fueron ECA y el 25% (15/60 casos, 25%) no fueron ECA (p.ej. ensayos de un solo grupo, tamaños de estudio pequeños, uso de resultados indirectos). Cuando los DO se reevaluaron en una evaluación estándar de beneficios, habiendo excedido el límite de ventas, la proporción de ECA aceptados y no ECA disminuyó al 65% (35/54 reevaluaciones) y al 13% (7/54 reevaluaciones), respectivamente. En 12 de las reevaluaciones de DO (22%), los estudios respectivos no fueron aceptados por el G-BA, lo que refleja los criterios de evidencia más estrictos aplicados en el proceso de evaluación estándar. El incumplimiento del ACT podría ser la razón principal de este cambio.
A diferencia de los DO, se aceptaron muy pocos ECA en las evaluaciones de los beneficios no relacionados con el DO (2/64 evaluaciones iniciales; 1/70 reevaluaciones). Por lo tanto, parece más probable que se acepten los ECA para los DO que para los que no lo son. Además, la evidencia del fabricante fue aceptada en todas las evaluaciones iniciales de DO, mientras que los estudios presentados fueron rechazados en 18 evaluaciones y reevaluaciones de no GO. Esto se debe al hecho de que en las evaluaciones de DO los estudios fundamentales siempre son aceptados por la ley.
Figura 1. Distribución del tipo de estudio entre DO y no DO en la evaluación inicial (AI) y la reevaluación (AR)
Llave: IA – evaluación inicial; No. –número; OD – medicamento huérfano; RA – reevaluación; ECA: ensayo controlado aleatorizado.
Nota: Seis DO que inicialmente se evaluaron como DO se reevaluaron como no DO debido a la pérdida de la condición de DO (*se agregaron cuatro SE anteriores; ** se redujeron en tres SE anteriores; *** se agregaron dos DO anteriores).
Además, y como era de esperar, hubo una correlación positiva entre un mayor tamaño de las poblaciones de estudio y la disponibilidad de un ECA (datos no mostrados).
Distribución de los beneficios añadidos concedidos para las DO y las no DO en la evaluación inicial
El grado de la prestación añadida concedida está vinculado a las pruebas aportadas. Cabe señalar que la concesión de "ningún beneficio adicional" no es aplicable a los OD en la evaluación de beneficios, y el grado predeterminado de beneficio médico adicional es "no cuantificable". La mayoría de las evaluaciones iniciales de DO dan como resultado un beneficio "no cuantificable" (Figura 2). En el caso de las personas que no son OD, la categoría de prestaciones más asignada fue "ninguna prestación añadida". La distribución de un beneficio añadido concedido en las categorías de "considerable" y "menor" era más o menos comparable en las evaluaciones iniciales de DO y no de DO.
Figura 2. Se le concedió un beneficio adicional en las evaluaciones iniciales

Figura 2. Se le concedió un beneficio adicional en las evaluaciones iniciales
Llave: No. –número; OD – medicamento huérfano.
Las evaluaciones de las prestaciones dan lugar a una disminución de los precios de reembolso (similar a los de los OD y a los que no lo son)
Un análisis de los niveles de precios antes de la evaluación del beneficio y después de la negociación del precio mostró una reducción del precio de lanzamiento mediante el procedimiento de una evaluación inicial (OD: 18% de descuento; no OD: 22% de descuento). Una nueva evaluación dio lugar a un nuevo aumento de la rebaja, aunque en menor medida (8% de descuento después de la reevaluación tanto para las DO como para las que no lo son). La diferencia de precio entre los OD y los que no lo son es grande. En promedio, se encontró que los DO eran aproximadamente de 10,2 a 13,5 veces más caros que los que no lo eran. Esta diferencia de costos persistió en el lanzamiento, después de la evaluación inicial de los beneficios y en las reevaluaciones (no se muestran los datos).

Medicamentos huérfanos: Equilibrio entre los costes elevados y las pruebas sólidas
El análisis mostró cómo se evalúan las DO, con precios típicamente más altos y pruebas menos sólidas que las que no lo son, en el sistema sanitario alemán. Si bien los DO constituyen una pequeña fracción de las evaluaciones de beneficios, sus gastos considerables, reflejados en los altos precios de reembolso en comparación con los que no lo son, subrayan su impacto significativo en el sistema de salud. El elevado costo de los DO está influenciado por múltiples factores, en particular la población restringida de pacientes y las importantes inversiones en investigación y desarrollo. En particular, los altos costos de los DO se discuten repetidamente en términos de si su precio está justificado en relación con la evidencia proporcionada, especialmente cuando se considera la carga económica en los sistemas de salud.
Los análisis indicaron comparabilidad en la evidencia presentada, y los ECA proporcionaron la base de evidencia para la mayoría de las evaluaciones tanto para DO como para no DO. Como muestran los datos, el estado de DO no está necesariamente asociado con la falta de calidad de la evidencia, que ha sido objeto de debates. Sin embargo, la reducción de los estudios aceptados para las DO en la reevaluación pone de relieve las dificultades de abordar el TCA. Como se anticipó, la baja prevalencia asociada con las enfermedades raras conduce a desafíos en la realización de ECA extensos, lo que destaca la correlación entre la prevalencia de la enfermedad y la calidad de la evidencia. Sin embargo, se demostró que las compañías farmacéuticas presentan pruebas de alta calidad, con un número significativo de ECA realizados para las DO, a pesar de las limitaciones mencionadas anteriormente.
En conclusión, los medicamentos desarrollados para enfermedades raras presentan distintos desafíos para los fabricantes y las autoridades de HTA. Si bien los "privilegios de evaluación" mejoran la disponibilidad de terapias para enfermedades raras difíciles de tratar, se justifica la precaución cuando se aplican criterios de medicina basada en la evidencia. Los resultados demuestran que, a pesar de los obstáculos a los que se enfrentan los DO, se proporciona evidencia de alta calidad en forma de ECA. Sin embargo, el sistema de ETS reconoce la inviabilidad ocasional de los ECA en el campo de las enfermedades raras, al tiempo que permite un mayor margen de maniobra para aceptar ECA no ECA en las evaluaciones de la DO.
Si bien este artículo no profundizó en la justificación de los altos precios de las DO, su correlación de eficacia o la justificación detrás del beneficio adicional otorgado, se justifica una evaluación adicional para analizar estos aspectos clave y su complejidad dentro del sistema de salud alemán. La interacción entre los costes de los DO, la solidez de la evidencia que ofrecen y el valor que aportan a los sistemas sanitarios sigue siendo un tema complejo y en evolución que requiere un escrutinio y análisis continuos.
En conclusión, los medicamentos desarrollados para enfermedades raras presentan distintos desafíos para los fabricantes y las autoridades de HTA. Si bien los "privilegios de evaluación" mejoran la disponibilidad de terapias para enfermedades raras difíciles de tratar, se justifica la precaución cuando se aplican criterios de medicina basada en la evidencia. Los resultados demuestran que, a pesar de los obstáculos a los que se enfrentan los DO, se proporciona evidencia de alta calidad en forma de ECA. Sin embargo, el sistema de ETS reconoce la inviabilidad ocasional de los ECA en el campo de las enfermedades raras, al tiempo que permite un mayor margen de maniobra para aceptar ECA no ECA en las evaluaciones de la DO.
Si bien este artículo no profundizó en la justificación de los altos precios de las DO, su correlación de eficacia o la justificación detrás del beneficio adicional otorgado, se justifica una evaluación adicional para analizar estos aspectos clave y su complejidad dentro del sistema de salud alemán. La interacción entre los costes de los DO, la solidez de la evidencia que ofrecen y el valor que aportan a los sistemas sanitarios sigue siendo un tema complejo y en evolución que requiere un escrutinio y análisis continuos.

Fuentes
-
Bundesministerium für Gesundheit (BMG). Seltene Erkrankungen. 2024. Consultado el 4 de septiembre de 2024. https://www.bundesgesundheitsministerium.de/themen/praevention/gesundheitsgefahren/seltene-erkrankungen.html
-
Bundesministerium der Justiz (BMJ). Sozialgesetzbuch (SGB) Fünftes Buch (V). Consultado el 4 de septiembre de 2024. https://www.gesetze-im-internet.de/sgb_5/__35a.html
-
Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen. Preis- und Kostenentwicklung von Orphan Drugs; Arbeitspapier [online]. 2024. Consultado el 19 de septiembre de 2024. https://dx.doi.org/10.60584/GA22-01


