Artikel
Aufdeckung der Evidenzlücke: Orphan Drugs vs. Non-Orphan Drugs in der deutschen Bewertung von Gesundheitstechnologien
-
Kristina Dittrich, PhD
-
Nadine Klusmeier, PhD
-
Mareike Kubinski, PhD
-

Jan-Frederik Löpmeier-Röh, MSc
-

Susanne Kossow, PhD
-
Michael Reinartz, PhD
-

Werner Kulp, PhD
Arzneimitteln für seltene Krankheiten wird oft vorgeworfen, dass es ihnen an Evidenz mangelt, weil sie einen gesetzlich garantierten Zusatznutzen haben. Aber ist dieses Vorurteil berechtigt? Dieser Artikel nimmt Sie mit auf eine aufschlussreiche Erkundung der Bewertung von Gesundheitstechnologien in Deutschland, um die Realität hinter diesen anhaltenden Annahmen zu verdeutlichen.

Seltene Erkrankungen und ihre Behandlung in der deutschen HTA
Seltene Krankheiten, von denen definitionsgemäß nur 1 von 2.000 Menschen in der Europäischen Union betroffen ist, haben oft keine wirksamen Behandlungsmöglichkeiten. Orphan Drugs (ODs) füllen diese Lücke und erhalten daher besondere Anreize für ihre Entwicklung und Kommerzialisierung. Für den Marktzugang in Deutschland erhalten ODs eine einzigartige Behandlung im Health Technology Assessment (HTA).
Die deutsche HTA für Arzneimittel ist im Arzneimittelmarktneuordnungsgesetz (AMNOG) geregelt. Das AMNOG zielt darauf ab, die Kostenerstattung für Arzneimittelhersteller und die Ausgabenbelastung für das nationale Gesundheitssystem in Einklang zu bringen. Arzneimittel sind verpflichtet, sich einer Nutzenbewertung gegenüber einer bestehenden Standardtherapie zu unterziehen (die sogenannte zweckmäßige Vergleichstherapie [ACT]). Eine Nutzenbewertung ist bei der Markteinführung eines neu zugelassenen Arzneimittels mit einem neuen Wirkstoff in Deutschland oder bei neu zugelassenen Indikationen eines bereits bewerteten Arzneimittels verpflichtend. Basierend auf der klinischen Evidenz bewertet der Gemeinsame Bundesausschuss (G-BA) den medizinischen Zusatznutzen anhand von sechs Kategorien ("groß", "erheblich", "gering", "gering", "nicht quantifizierbar", "kein Zusatznutzen" oder "weniger").
Für ODs wird ein medizinischer Zusatznutzen gesetzlich gewährt, und die Beurteilung des Ausmaßes basiert auf entscheidenden Nachweisen, ohne dass ein Vergleich mit einem ACT erforderlich ist. Wenn der Umsatz jedoch 30 Millionen Euro erreicht oder die Indikation den Status eines Orphan verliert, ist eine Neubewertung im Rahmen des Standardbewertungsverfahrens obligatorisch. Non-Orphan Drugs (Non-ODs), die zur regelmäßigen Nutzenbewertung verpflichtet sind, können ebenfalls einer Neubewertung unterzogen werden, entweder zwingend bei vorläufigen Entscheidungen (z.B. werden zusätzliche Daten vom G-BA angefordert) oder auf Antrag des Arzneimittelherstellers.
Die Nutzenbewertung ist die Grundlage für die Preisverhandlung für die Kostenerstattung zwischen dem Arzneimittelhersteller und dem GKV-SV-Spitzenverband. Nur Arzneimittel mit medizinischem Zusatznutzen können in der Preisverhandlung einen höheren Preis erzielen als die Kosten für die ACT. Innerhalb der ersten sechs Monate nach der Markteinführung kann der Hersteller den Einführungspreis frei festlegen, während der ausgehandelte Preis ab dem siebten Monat angewendet wird. Der Erstattungspreis für ein Medikament kann sich mit der Zeit weiter ändern, wenn Neubewertungen und neue Indikationen für das jeweilige Medikament die HTA und die anschließenden Preisverhandlungen durchlaufen.
Dieser Artikel untersucht die Ergebnisse einer eingehenden Analyse deutscher HTAs, die sich auf den Vergleich der Evidenzkriterien und der Preisgestaltung zwischen ODs und Nicht-ODs (sowohl für die Erstbewertung als auch für die Neubewertung) konzentriert, und zielt darauf ab, zu beleuchten, ob ODs im Vergleich zu Nicht-ODs keine Evidenz in HTA aufweisen.
In einer umfassenden Übersichtsarbeit wurden Nutzenbewertungen für ODs und Nicht-ODs von 2011 bis Mai 2024 aus einer Datenbank mit Daten des G-BA analysiert. Diese Analyse umfasste 959 Nutzenbewertungen. Davon handelte es sich bei 810 um Erstbewertungen von neu zugelassenen Arzneimitteln oder neu zugelassenen Indikationen eines Arzneimittels, während es sich bei 149 um Neubewertungen handelte, wobei einige Produkte für eine jeweilige Indikation mehr als einmal neu bewertet wurden. Die Analyse von 810 anfänglichen Nutzenbewertungen ergab, dass 27 % der Bewertungen ODs bewerteten, während 73 % Nicht-ODs waren. Insgesamt wurde für 15 % aller Bewertungen (7 % ODs und 8 % Non-ODs) eine Neubewertung durchgeführt.
Alle Neubewertungen wurden, falls zutreffend, mit der jeweiligen ursprünglichen Nutzenbewertung abgeglichen, um verwandte Bewertungen für dasselbe Medikament aufeinander abzustimmen. Die Daten wurden für ODs und Nicht-ODs extrahiert, wobei der Schwerpunkt auf Parametern wie der bewerteten Evidenz (Studientypen, Anzahl der Studien und Größe der Studien), dem Grad des gewährten Zusatznutzens und dem Preis lag. Die Evaluierungen konzentrierten sich auf Fälle mit einer Erstbewertung (60 ODs und 64 Nicht-ODs) und einer Neubewertung (54 ODs und 70 Nicht-ODs, was widerspiegelt, dass einige Arzneimittel den OD-Status aus der Prävalenzperspektive verloren und nach dem Standardverfahren als Nicht-ODs neu bewertet wurden) für eine bestimmte Indikation, um zu beurteilen, ob sich die Evidenz, das Nutzenniveau und der Preis zwischen der Erstbewertung und der Neubewertung geändert haben. und zum Vergleich zwischen ODs und Nicht-ODs.
Für ODs wird ein medizinischer Zusatznutzen gesetzlich gewährt, und die Beurteilung des Ausmaßes basiert auf entscheidenden Nachweisen, ohne dass ein Vergleich mit einem ACT erforderlich ist. Wenn der Umsatz jedoch 30 Millionen Euro erreicht oder die Indikation den Status eines Orphan verliert, ist eine Neubewertung im Rahmen des Standardbewertungsverfahrens obligatorisch. Non-Orphan Drugs (Non-ODs), die zur regelmäßigen Nutzenbewertung verpflichtet sind, können ebenfalls einer Neubewertung unterzogen werden, entweder zwingend bei vorläufigen Entscheidungen (z.B. werden zusätzliche Daten vom G-BA angefordert) oder auf Antrag des Arzneimittelherstellers.
Die Nutzenbewertung ist die Grundlage für die Preisverhandlung für die Kostenerstattung zwischen dem Arzneimittelhersteller und dem GKV-SV-Spitzenverband. Nur Arzneimittel mit medizinischem Zusatznutzen können in der Preisverhandlung einen höheren Preis erzielen als die Kosten für die ACT. Innerhalb der ersten sechs Monate nach der Markteinführung kann der Hersteller den Einführungspreis frei festlegen, während der ausgehandelte Preis ab dem siebten Monat angewendet wird. Der Erstattungspreis für ein Medikament kann sich mit der Zeit weiter ändern, wenn Neubewertungen und neue Indikationen für das jeweilige Medikament die HTA und die anschließenden Preisverhandlungen durchlaufen.
Dieser Artikel untersucht die Ergebnisse einer eingehenden Analyse deutscher HTAs, die sich auf den Vergleich der Evidenzkriterien und der Preisgestaltung zwischen ODs und Nicht-ODs (sowohl für die Erstbewertung als auch für die Neubewertung) konzentriert, und zielt darauf ab, zu beleuchten, ob ODs im Vergleich zu Nicht-ODs keine Evidenz in HTA aufweisen.
In einer umfassenden Übersichtsarbeit wurden Nutzenbewertungen für ODs und Nicht-ODs von 2011 bis Mai 2024 aus einer Datenbank mit Daten des G-BA analysiert. Diese Analyse umfasste 959 Nutzenbewertungen. Davon handelte es sich bei 810 um Erstbewertungen von neu zugelassenen Arzneimitteln oder neu zugelassenen Indikationen eines Arzneimittels, während es sich bei 149 um Neubewertungen handelte, wobei einige Produkte für eine jeweilige Indikation mehr als einmal neu bewertet wurden. Die Analyse von 810 anfänglichen Nutzenbewertungen ergab, dass 27 % der Bewertungen ODs bewerteten, während 73 % Nicht-ODs waren. Insgesamt wurde für 15 % aller Bewertungen (7 % ODs und 8 % Non-ODs) eine Neubewertung durchgeführt.
Alle Neubewertungen wurden, falls zutreffend, mit der jeweiligen ursprünglichen Nutzenbewertung abgeglichen, um verwandte Bewertungen für dasselbe Medikament aufeinander abzustimmen. Die Daten wurden für ODs und Nicht-ODs extrahiert, wobei der Schwerpunkt auf Parametern wie der bewerteten Evidenz (Studientypen, Anzahl der Studien und Größe der Studien), dem Grad des gewährten Zusatznutzens und dem Preis lag. Die Evaluierungen konzentrierten sich auf Fälle mit einer Erstbewertung (60 ODs und 64 Nicht-ODs) und einer Neubewertung (54 ODs und 70 Nicht-ODs, was widerspiegelt, dass einige Arzneimittel den OD-Status aus der Prävalenzperspektive verloren und nach dem Standardverfahren als Nicht-ODs neu bewertet wurden) für eine bestimmte Indikation, um zu beurteilen, ob sich die Evidenz, das Nutzenniveau und der Preis zwischen der Erstbewertung und der Neubewertung geändert haben. und zum Vergleich zwischen ODs und Nicht-ODs.

Vergleichbarkeit der vorgelegten Nachweise für ODs und Nicht-ODs
Für ODs wird automatisch ein Zusatznutzen per Gesetz gewährt. Welche klinische Evidenz wird daher für ODs in Nutzenbewertungen vorgelegt und wie unterscheidet sie sich von Nicht-OD-Bewertungen? In einem ersten Schritt der Standardnutzenbewertung bewertet der G-BA die Qualität der eingereichten Studien anhand des Evidenzgrades, um einen hinreichend belastbaren Kenntnisstand zu gewährleisten. Basierend auf dieser Bewertung wird die Studie entweder angenommen oder nicht in der Bewertung berücksichtigt. Darüber hinaus müssen Studien, die für eine Standardnutzenbewertung vorgesehen sind, einen Vergleich des Arzneimittels mit dem vom G-BA definierten ACT beinhalten. Während ODs von einer weniger strengen Ausführung von Evidenzkriterien für Erstbewertungen profitieren, werden diejenigen, die die Verkaufsgrenze überschreiten, nach dem Standardverfahren neu bewertet, auch wenn sie aus Sicht der Prävalenz den Status eines verwaisten Leidens behalten.
Die Auswertungen zeigten, dass randomisierte kontrollierte Studien (RCTs), der Goldstandard der Evidenz, sowohl für ODs als auch für Nicht-ODs der vorherrschende Studientyp waren (Abbildung 1). Bei Nicht-ODs wurden vom G-BA mehr RCTs in der Neubewertung akzeptiert (51/70 Fälle, 73 %) als in der Erstbewertung (44/64 Fälle, 69 %), während es bei den ODs umgekehrt war (45/60 Erstbewertungen, 75 %; 35/54 Neubewertungen, 65 %).
Zunächst wurden die vom Hersteller vorgelegten klinischen Studien vom G-BA in allen OD-Bewertungen akzeptiert; die Mehrzahl der Studien (45/60 Fälle, 75 %) waren RCTs und 25 % (15/60 Fälle, 25 %) waren Nicht-RCTs (z. B. einarmige Studien, kleine Studiengrößen, Verwendung von Surrogat-Endpunkten). Bei einer Neubewertung der ODs in einer Standardnutzenbewertung sank der Anteil der akzeptierten RCTs und Nicht-RCTs auf 65 % (35 von 54 Neubewertungen) bzw. 13 % (7 von 54 Neubewertungen). In 12 der OD-Reassessments (22%) wurden die entsprechenden Studien vom G-BA nicht akzeptiert, was die verschärften Evidenzkriterien des Standardbewertungsverfahrens widerspiegelt. Die Nichteinhaltung des ACT könnte der Hauptgrund für diese Verschiebung sein.
Im Gegensatz zu ODs wurden nur sehr wenige Nicht-RCTs in Nicht-OD-Nutzenbewertungen akzeptiert (2/64 Erstbewertungen; 1/70 Rebewertungen). Daher scheinen Nicht-RCTs für ODs eher akzeptiert zu werden als für Nicht-ODs. Darüber hinaus wurden die Nachweise des Herstellers in allen OD-Erstbewertungen akzeptiert, während die vorgelegten Studien in 18 Bewertungen und Neubewertungen von Nicht-ODs zurückgewiesen wurden. Dies ist darauf zurückzuführen, dass bei OD-Assessments immer zulassungsrelevante Studien gesetzlich anerkannt werden.
Abbildung 1. Verteilung des Studientyps auf ODs und Nicht-ODs in der Erstbewertung (IA) und Neubewertung (RA)
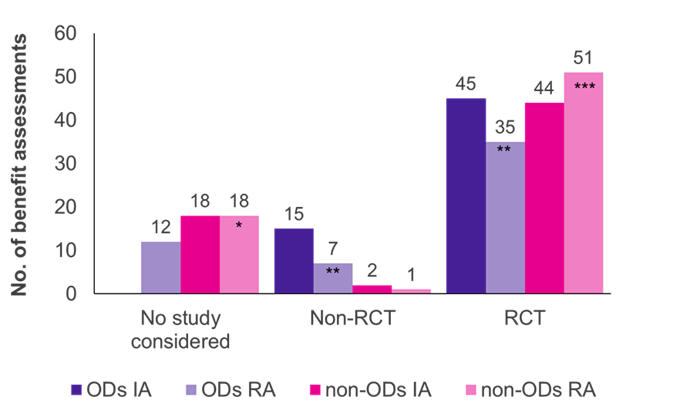
Schlüssel: Folgenabschätzung – Erstbewertung; Nein. –Zahl; OD – Orphan Drug; RA – Neubewertung; RCT – randomisierte kontrollierte Studie.
Darüber hinaus gab es, und wenig überraschend, eine positive Korrelation zwischen einer erhöhten Größe der Studienpopulationen und der Verfügbarkeit einer RCT (Daten nicht gezeigt).
Zunächst wurden die vom Hersteller vorgelegten klinischen Studien vom G-BA in allen OD-Bewertungen akzeptiert; die Mehrzahl der Studien (45/60 Fälle, 75 %) waren RCTs und 25 % (15/60 Fälle, 25 %) waren Nicht-RCTs (z. B. einarmige Studien, kleine Studiengrößen, Verwendung von Surrogat-Endpunkten). Bei einer Neubewertung der ODs in einer Standardnutzenbewertung sank der Anteil der akzeptierten RCTs und Nicht-RCTs auf 65 % (35 von 54 Neubewertungen) bzw. 13 % (7 von 54 Neubewertungen). In 12 der OD-Reassessments (22%) wurden die entsprechenden Studien vom G-BA nicht akzeptiert, was die verschärften Evidenzkriterien des Standardbewertungsverfahrens widerspiegelt. Die Nichteinhaltung des ACT könnte der Hauptgrund für diese Verschiebung sein.
Im Gegensatz zu ODs wurden nur sehr wenige Nicht-RCTs in Nicht-OD-Nutzenbewertungen akzeptiert (2/64 Erstbewertungen; 1/70 Rebewertungen). Daher scheinen Nicht-RCTs für ODs eher akzeptiert zu werden als für Nicht-ODs. Darüber hinaus wurden die Nachweise des Herstellers in allen OD-Erstbewertungen akzeptiert, während die vorgelegten Studien in 18 Bewertungen und Neubewertungen von Nicht-ODs zurückgewiesen wurden. Dies ist darauf zurückzuführen, dass bei OD-Assessments immer zulassungsrelevante Studien gesetzlich anerkannt werden.
Abbildung 1. Verteilung des Studientyps auf ODs und Nicht-ODs in der Erstbewertung (IA) und Neubewertung (RA)
Schlüssel: Folgenabschätzung – Erstbewertung; Nein. –Zahl; OD – Orphan Drug; RA – Neubewertung; RCT – randomisierte kontrollierte Studie.
Anmerkung: Sechs ODs, die ursprünglich als ODs bewertet wurden, wurden aufgrund des Verlusts des OD-Status als Nicht-OD neu bewertet (*vier ehemalige ODs hinzugefügt; **reduziert um drei ehemalige ODs; ***zwei ehemalige ODs hinzugefügt).
Darüber hinaus gab es, und wenig überraschend, eine positive Korrelation zwischen einer erhöhten Größe der Studienpopulationen und der Verfügbarkeit einer RCT (Daten nicht gezeigt).
Verteilung des gewährten Zusatznutzens für ODs und Nicht-ODs in der Ersteinschätzung
Die Höhe des gewährten Zusatznutzens hängt von den vorgelegten Nachweisen ab. Bemerkenswert ist, dass die Gewährung "kein Zusatznutzen" für ODs in der Nutzenbewertung nicht anwendbar ist und der Standardgrad des medizinischen Zusatznutzens "nicht quantifizierbar" ist. Die meisten OD-Erstbewertungen führen zu einem "nicht quantifizierbaren" Nutzen (Abbildung 2). Für Nicht-ODs war die am häufigsten zugewiesene Leistungskategorie "kein Zusatznutzen". Die Verteilung eines gewährten Zusatznutzens in die Kategorien "erheblich" und "gering" war bei den Ersteinschätzungen für die Effektivität und die Nicht-Effektivität mehr oder weniger vergleichbar.
Abbildung 2. Gewährter Zusatznutzen bei Ersteinschätzungen

Abbildung 2. Gewährter Zusatznutzen bei Ersteinschätzungen
Schlüssel: Nein. –Zahl; OD – Orphan Drug.
Nutzenbewertungen führen zu einer Senkung der Erstattungspreise (ähnlich wie bei ODs und Non-ODs)
Eine Analyse des Preisniveaus vor der Nutzenbewertung und nach der Preisverhandlung ergab eine Senkung des Einführungspreises durch das Verfahren einer Erstbewertung (OD: 18 % Rabatt; Nicht-OD: 22 % Rabatt). Eine Neubewertung führte zu einer weiteren Erhöhung des Preisnachlasses, wenn auch in geringerem Umfang (8 % Rabatt nach Neubewertung sowohl für OE als auch für Nicht-OD). Der Preisunterschied zwischen ODs und Nicht-ODs ist groß. Im Durchschnitt waren ODs etwa 10,2 bis 13,5 Mal teurer als Nicht-ODs. Dieser Kostenunterschied blieb bei der Einführung, nach der ersten Nutzenbewertung und über Neubewertungen hinweg bestehen (Daten nicht dargestellt).

Orphan Drugs: Abwägung zwischen hohen Kosten und aussagekräftigen Beweisen
Die Analyse zeigte, wie ODs im deutschen Gesundheitssystem mit typischerweise höheren Preisen und weniger robuster Evidenz als Non-ODs bewertet werden. ODs machen zwar nur einen kleinen Teil der Nutzenbewertungen aus, aber ihre erheblichen Kosten, die sich in den hohen Erstattungspreisen im Vergleich zu Nicht-ODs widerspiegeln, unterstreichen ihre erheblichen Auswirkungen auf das Gesundheitssystem. Die erhöhten Kosten für ODs werden von mehreren Faktoren beeinflusst, insbesondere von der begrenzten Patientenpopulation und den erheblichen Investitionen in Forschung und Entwicklung. Insbesondere die hohen Kosten von ODs werden immer wieder im Hinblick darauf diskutiert, ob ihr Preis im Verhältnis zu den vorgelegten Nachweisen gerechtfertigt ist, insbesondere unter Berücksichtigung der wirtschaftlichen Belastung der Gesundheitssysteme.
Die Analysen zeigten eine Vergleichbarkeit der vorgelegten Evidenz, wobei RCTs die Evidenzbasis für die Mehrzahl der Bewertungen sowohl für ODs als auch für Nicht-ODs bildeten. Wie aus den Daten hervorgeht, ist der OD-Status nicht notwendigerweise mit einer mangelnden Qualität der Evidenz verbunden, die Gegenstand von Debatten war. Der Rückgang der in der Neubewertung akzeptierten Studien für OD unterstreicht jedoch die Schwierigkeiten bei der Bewältigung der ACT. Wie erwartet, führt die geringe Prävalenz im Zusammenhang mit seltenen Krankheiten zu Herausforderungen bei der Durchführung umfangreicher RCTs, die den Zusammenhang zwischen der Krankheitsprävalenz und der Qualität der Evidenz hervorheben. Es zeigte sich jedoch, dass Pharmaunternehmen trotz der oben genannten Einschränkungen qualitativ hochwertige Evidenz vorlegen, wobei eine beträchtliche Anzahl von RCTs für ODs durchgeführt wurde.
Zusammenfassend lässt sich sagen, dass Medikamente, die für seltene Krankheiten entwickelt wurden, erhebliche Herausforderungen für Hersteller und HTA-Behörden darstellen. Während "Bewertungsprivilegien" die Verfügbarkeit von Therapien für schwer zu behandelnde seltene Erkrankungen verbessern, ist Vorsicht geboten, wenn evidenzbasierte medizinische Kriterien angewendet werden. Unsere Ergebnisse zeigen, dass trotz der Hindernisse, mit denen ODs konfrontiert sind, qualitativ hochwertige Evidenz in Form von RCTs erbracht wird. Das HTA-System erkennt jedoch die gelegentliche Undurchführbarkeit von RCTs im Bereich der seltenen Krankheiten an und lässt gleichzeitig mehr Spielraum bei der Akzeptanz von Nicht-RCTs in OD-Bewertungen.
Obwohl in diesem Artikel nicht auf die Rechtfertigung der hohen Preise für ODs, ihre Wirksamkeitskorrelation oder die Gründe für den gewährten Zusatznutzen eingegangen wurde, ist eine weitere Evaluierung erforderlich, um diese Schlüsselaspekte und ihre Komplexität innerhalb des deutschen Gesundheitssystems zu analysieren. Das Zusammenspiel zwischen den Kosten von ODs, der Stärke der Evidenz, die sie bieten, und dem Nutzen, den sie für die Gesundheitssysteme bringen, bleibt ein komplexes und sich entwickelndes Thema, das eine kontinuierliche Prüfung und Analyse erfordert.
Zusammenfassend lässt sich sagen, dass Medikamente, die für seltene Krankheiten entwickelt wurden, erhebliche Herausforderungen für Hersteller und HTA-Behörden darstellen. Während "Bewertungsprivilegien" die Verfügbarkeit von Therapien für schwer zu behandelnde seltene Erkrankungen verbessern, ist Vorsicht geboten, wenn evidenzbasierte medizinische Kriterien angewendet werden. Unsere Ergebnisse zeigen, dass trotz der Hindernisse, mit denen ODs konfrontiert sind, qualitativ hochwertige Evidenz in Form von RCTs erbracht wird. Das HTA-System erkennt jedoch die gelegentliche Undurchführbarkeit von RCTs im Bereich der seltenen Krankheiten an und lässt gleichzeitig mehr Spielraum bei der Akzeptanz von Nicht-RCTs in OD-Bewertungen.
Obwohl in diesem Artikel nicht auf die Rechtfertigung der hohen Preise für ODs, ihre Wirksamkeitskorrelation oder die Gründe für den gewährten Zusatznutzen eingegangen wurde, ist eine weitere Evaluierung erforderlich, um diese Schlüsselaspekte und ihre Komplexität innerhalb des deutschen Gesundheitssystems zu analysieren. Das Zusammenspiel zwischen den Kosten von ODs, der Stärke der Evidenz, die sie bieten, und dem Nutzen, den sie für die Gesundheitssysteme bringen, bleibt ein komplexes und sich entwickelndes Thema, das eine kontinuierliche Prüfung und Analyse erfordert.

Quellen
-
Bundesministerium für Gesundheit (BMG). Seltene Erkrankungen. 2024. Abgerufen am 4. September 2024. https://www.bundesgesundheitsministerium.de/themen/praevention/gesundheitsgefahren/seltene-erkrankungen.html
-
Bundesministerium der Justiz (BMJ). Sozialgesetzbuch (SGB) Fünftes Buch (V). Abgerufen am 4. September 2024. https://www.gesetze-im-internet.de/sgb_5/__35a.html
-
Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen. Preis- und Kostenentwicklung von Orphan Drugs; Arbeitspapier [online]. 2024. Abgerufen am 19. September 2024. https://dx.doi.org/10.60584/GA22-01


