Article
Dévoiler l’écart en matière de données probantes : Médicaments orphelins et médicaments non orphelins dans les évaluations allemandes des technologies de la santé
-
Kristina Dittrich, PhD
-
Nadine Klusmeier, PhD
-
Mareike Kubinski, PhD
-

Jan-Frederik Löpmeier-Röh, MSc
-

Susanne Kossow, PhD
-
Michael Reinartz, PhD
-

Werner Kulp, PhD
Les médicaments pour les maladies rares sont souvent préjugés manquer de preuves parce qu’ils ont un avantage supplémentaire garanti par la loi. Mais ce préjugé est-il justifié ? Cet article vous emmène dans une exploration éclairante de l’évaluation des technologies de la santé en Allemagne afin d’élucider la réalité derrière ces hypothèses persistantes.

Les maladies rares et leurs traitements en ETS allemande
Les maladies rares, qui ne touchent par définition qu’une personne sur 2 000 dans l’Union européenne, manquent souvent d’options de traitement efficaces. Les médicaments orphelins comblent cette lacune et bénéficient donc d’incitations spéciales pour leur développement et leur commercialisation. Pour l’accès au marché en Allemagne, les DO reçoivent un traitement unique dans le domaine de l’évaluation des technologies de la santé (ETS).
L’ETS allemande pour les médicaments est réglementée par l’Arzneimittelmarktneuordnungsgesetz (loi sur la réorganisation du marché pharmaceutique, AMNOG). L’AMNOG vise à équilibrer le remboursement des fabricants de produits pharmaceutiques et la charge des dépenses pour le système de santé national. Les médicaments doivent faire l’objet d’une évaluation de leur bénéfice par rapport à un traitement standard existant (ce que l’on appelle le traitement [ACT]comparatif approprié). Une évaluation du bénéfice est obligatoire lors du lancement d’un médicament nouvellement autorisé contenant une nouvelle substance active en Allemagne ou pour les indications nouvellement autorisées d’un médicament déjà évalué. Sur la base des preuves cliniques, le Comité mixte fédéral (G-BA) évalue le bénéfice médical supplémentaire à l’aide de six catégories (« majeur », « considérable », « mineur », « non quantifiable », « aucun avantage ajouté » ou « moins »).
Pour les OD, une prestation médicale supplémentaire est accordée par la loi, et l’évaluation de l’étendue est basée sur des preuves cruciales sans obligation de comparaison avec un ACT. Toutefois, si le chiffre d’affaires atteint 30 millions d’euros ou si l’indication perd son statut d’orpheline, une réévaluation dans le cadre du processus d’évaluation standard devient obligatoire. Les médicaments non orphelins (non-OD), qui sont soumis à l’évaluation régulière des prestations, peuvent également faire l’objet d’une réévaluation, soit de manière obligatoire en cas de décisions temporaires (par exemple, des données supplémentaires sont demandées par le G-BA), soit à la demande du fabricant de produits pharmaceutiques.
L’évaluation des prestations sert de base à la négociation du prix de remboursement entre le fabricant de produits pharmaceutiques et l’Association nationale des caisses obligatoires d’assurance maladie (GKV-SV). Seuls les médicaments présentant un avantage médical supplémentaire peuvent obtenir un prix plus élevé que les coûts de l’ACT lors de la négociation des prix. Dans les six premiers mois suivant le lancement, le fabricant est libre de définir le prix de lancement, tandis que le prix négocié est appliqué à partir du septième mois. Le prix de remboursement d’un médicament peut encore changer avec le temps, lorsque les réévaluations et les nouvelles indications pour le médicament concerné passent par l’ETS et les négociations de prix ultérieures.
Cet article explore les résultats d’une analyse approfondie des ETS allemandes centrée sur la comparaison des critères de preuve et des prix entre les DO et les non-OD (à la fois pour l’évaluation initiale et la réévaluation) et vise à faire la lumière sur la question de savoir si les OD manquent de preuves dans les ETS par rapport aux non-OD.
Un examen approfondi a analysé les évaluations des avantages pour les OD et les non-OD de 2011 à mai 2024 à partir d’une base de données de G-BA. Cette analyse a porté sur 959 évaluations des prestations. Parmi ceux-ci, 810 étaient des évaluations initiales de médicaments nouvellement autorisés ou d’indications nouvellement autorisées d’un médicament, tandis que 149 étaient des réévaluations, certains produits étant réévalués plus d’une fois pour une indication respective. L’analyse de 810 évaluations initiales des prestations a révélé que 27 % des évaluations évaluaient les DO, tandis que 73 % étaient des non-OD. Au total, une réévaluation a été effectuée pour 15 % de toutes les évaluations (7 % de DO et 8 % de non-OD).
Toute réévaluation a été appariée à l’évaluation initiale des avantages respectifs, le cas échéant, afin d’harmoniser les évaluations connexes pour le même médicament. Les données ont été extraites pour les DO et les non-DO en se concentrant sur des paramètres tels que les données probantes évaluées (types d’études, nombre d’études et taille des études), le niveau d’avantage supplémentaire accordé et le prix. Les évaluations se sont concentrées sur les cas comportant à la fois une évaluation initiale (60 DO et 64 non-OD) et une réévaluation (54 DO et 70 non-OD, reflétant le fait que certains médicaments ont perdu le statut de DO du point de vue de la prévalence et ont été réévalués selon la procédure standard en tant que non-OD) pour une indication donnée afin d’évaluer si les données probantes, le niveau de bénéfice et le prix ont changé entre l’évaluation initiale et la réévaluation. et de comparer entre les OD et les non-OD.
Pour les OD, une prestation médicale supplémentaire est accordée par la loi, et l’évaluation de l’étendue est basée sur des preuves cruciales sans obligation de comparaison avec un ACT. Toutefois, si le chiffre d’affaires atteint 30 millions d’euros ou si l’indication perd son statut d’orpheline, une réévaluation dans le cadre du processus d’évaluation standard devient obligatoire. Les médicaments non orphelins (non-OD), qui sont soumis à l’évaluation régulière des prestations, peuvent également faire l’objet d’une réévaluation, soit de manière obligatoire en cas de décisions temporaires (par exemple, des données supplémentaires sont demandées par le G-BA), soit à la demande du fabricant de produits pharmaceutiques.
L’évaluation des prestations sert de base à la négociation du prix de remboursement entre le fabricant de produits pharmaceutiques et l’Association nationale des caisses obligatoires d’assurance maladie (GKV-SV). Seuls les médicaments présentant un avantage médical supplémentaire peuvent obtenir un prix plus élevé que les coûts de l’ACT lors de la négociation des prix. Dans les six premiers mois suivant le lancement, le fabricant est libre de définir le prix de lancement, tandis que le prix négocié est appliqué à partir du septième mois. Le prix de remboursement d’un médicament peut encore changer avec le temps, lorsque les réévaluations et les nouvelles indications pour le médicament concerné passent par l’ETS et les négociations de prix ultérieures.
Cet article explore les résultats d’une analyse approfondie des ETS allemandes centrée sur la comparaison des critères de preuve et des prix entre les DO et les non-OD (à la fois pour l’évaluation initiale et la réévaluation) et vise à faire la lumière sur la question de savoir si les OD manquent de preuves dans les ETS par rapport aux non-OD.
Un examen approfondi a analysé les évaluations des avantages pour les OD et les non-OD de 2011 à mai 2024 à partir d’une base de données de G-BA. Cette analyse a porté sur 959 évaluations des prestations. Parmi ceux-ci, 810 étaient des évaluations initiales de médicaments nouvellement autorisés ou d’indications nouvellement autorisées d’un médicament, tandis que 149 étaient des réévaluations, certains produits étant réévalués plus d’une fois pour une indication respective. L’analyse de 810 évaluations initiales des prestations a révélé que 27 % des évaluations évaluaient les DO, tandis que 73 % étaient des non-OD. Au total, une réévaluation a été effectuée pour 15 % de toutes les évaluations (7 % de DO et 8 % de non-OD).
Toute réévaluation a été appariée à l’évaluation initiale des avantages respectifs, le cas échéant, afin d’harmoniser les évaluations connexes pour le même médicament. Les données ont été extraites pour les DO et les non-DO en se concentrant sur des paramètres tels que les données probantes évaluées (types d’études, nombre d’études et taille des études), le niveau d’avantage supplémentaire accordé et le prix. Les évaluations se sont concentrées sur les cas comportant à la fois une évaluation initiale (60 DO et 64 non-OD) et une réévaluation (54 DO et 70 non-OD, reflétant le fait que certains médicaments ont perdu le statut de DO du point de vue de la prévalence et ont été réévalués selon la procédure standard en tant que non-OD) pour une indication donnée afin d’évaluer si les données probantes, le niveau de bénéfice et le prix ont changé entre l’évaluation initiale et la réévaluation. et de comparer entre les OD et les non-OD.

Comparabilité des données probantes présentées pour les DO et les non-DO
Pour les OD, un avantage supplémentaire est automatiquement accordé par la loi. Par conséquent, quelles sont les preuves cliniques présentées pour les DO dans les évaluations des avantages, et en quoi diffèrent-elles des évaluations autres que les DO ? Dans un premier temps, dans le cadre de l’évaluation standard des prestations, le G-BA évalue la qualité des études soumises en fonction du niveau de preuve afin de fournir un niveau de connaissances suffisamment fiable. Sur la base de cette évaluation, l’étude est acceptée ou n’est pas prise en compte dans l’évaluation. De plus, les études fournies pour une évaluation standard des bénéfices doivent inclure une comparaison du médicament avec l’ACT, défini par le G-BA. Alors que les DO bénéficient d’une exécution moins stricte des critères de preuve pour les évaluations initiales, celles qui dépassent la limite de vente sont réévaluées selon la procédure standard, même si elles conservent leur statut d’orphelin du point de vue de la prévalence.
Les évaluations ont montré que les essais contrôlés randomisés (ECR), l’étalon-or des preuves, étaient le type d’étude prédominant pour les DO et les non-DO (Figure 1). En ce qui concerne les non-OD, un plus grand nombre d’ECR ont été acceptés par le G-BA lors de la réévaluation (51/70 cas, 73 %) que dans le cadre de l’évaluation initiale (44/64 cas, 69 %), alors que c’était l’inverse pour les OD (45/60 évaluations initiales, 75 % ; 35/54 réévaluations, 65 %).
Initialement, les études cliniques soumises par le fabricant ont été acceptées par le G-BA dans toutes les évaluations de la DO ; la majorité des études (45/60 cas, 75 %) étaient des ECR, et 25 % (15/60 cas, 25 %) n’étaient pas des ECR (p. ex., essais à un seul bras, études de petite taille, utilisation de résultats de substitution). Lorsque les DO ont été réévalués dans le cadre d’une évaluation standard des avantages, après avoir dépassé la limite de vente, la proportion d’ECR acceptés et de non-ECR a diminué à 65 % (réévaluations 35/54) et à 13 % (réévaluations 7/54), respectivement. Dans 12 des réévaluations de la DO (22 %), les études respectives n’ont pas été acceptées par le G-BA, ce qui reflète les critères de preuve plus stricts appliqués dans le cadre du processus d’évaluation standard. Le non-respect de l’ACT pourrait être la principale raison de ce changement.
Contrairement aux DO, très peu de non-ECR ont été acceptés dans les évaluations des prestations autres que les DO (2/64 évaluations initiales ; 1/70 réévaluations). Ainsi, les non-ECR semblent plus susceptibles d’être acceptés pour les DO que pour les non-OD. De plus, les données probantes du fabricant ont été acceptées dans toutes les évaluations initiales des DO, alors que les études présentées ont été rejetées dans 18 évaluations et réévaluations de non-OD. Cela est dû au fait que, dans les évaluations de la DO, les études pivots sont toujours acceptées par la loi.
Graphique 1. Répartition du type d’étude entre les DO et les non-DO dans l’évaluation initiale (AI) et la réévaluation (RA)
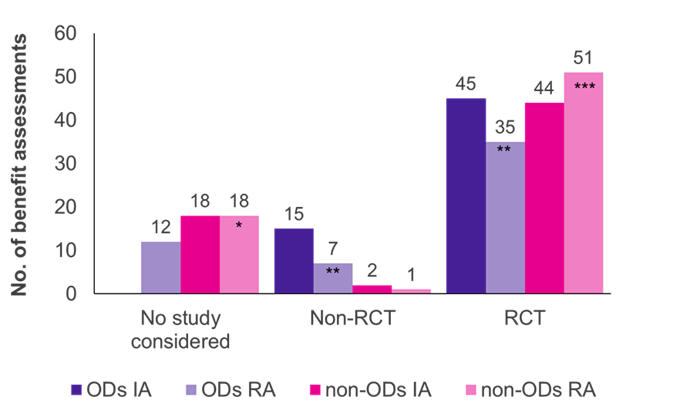
Clé: AI – évaluation initiale ; Non. –nombre; OD – médicament orphelin ; PR – réévaluation ; ECR – essai contrôlé randomisé.
De plus, et sans surprise, il y avait une corrélation positive entre l’augmentation de la taille des populations étudiées et la disponibilité d’un ECR (données non présentées).
Initialement, les études cliniques soumises par le fabricant ont été acceptées par le G-BA dans toutes les évaluations de la DO ; la majorité des études (45/60 cas, 75 %) étaient des ECR, et 25 % (15/60 cas, 25 %) n’étaient pas des ECR (p. ex., essais à un seul bras, études de petite taille, utilisation de résultats de substitution). Lorsque les DO ont été réévalués dans le cadre d’une évaluation standard des avantages, après avoir dépassé la limite de vente, la proportion d’ECR acceptés et de non-ECR a diminué à 65 % (réévaluations 35/54) et à 13 % (réévaluations 7/54), respectivement. Dans 12 des réévaluations de la DO (22 %), les études respectives n’ont pas été acceptées par le G-BA, ce qui reflète les critères de preuve plus stricts appliqués dans le cadre du processus d’évaluation standard. Le non-respect de l’ACT pourrait être la principale raison de ce changement.
Contrairement aux DO, très peu de non-ECR ont été acceptés dans les évaluations des prestations autres que les DO (2/64 évaluations initiales ; 1/70 réévaluations). Ainsi, les non-ECR semblent plus susceptibles d’être acceptés pour les DO que pour les non-OD. De plus, les données probantes du fabricant ont été acceptées dans toutes les évaluations initiales des DO, alors que les études présentées ont été rejetées dans 18 évaluations et réévaluations de non-OD. Cela est dû au fait que, dans les évaluations de la DO, les études pivots sont toujours acceptées par la loi.
Graphique 1. Répartition du type d’étude entre les DO et les non-DO dans l’évaluation initiale (AI) et la réévaluation (RA)
Clé: AI – évaluation initiale ; Non. –nombre; OD – médicament orphelin ; PR – réévaluation ; ECR – essai contrôlé randomisé.
Note: Six OD, initialement évalués comme OD, ont été réévalués comme non-OD en raison de la perte du statut de DO (*ajout de quatre anciens OD ; **réduit de trois anciens OD ; ***ajout de deux anciens OD).
De plus, et sans surprise, il y avait une corrélation positive entre l’augmentation de la taille des populations étudiées et la disponibilité d’un ECR (données non présentées).
Répartition des prestations supplémentaires accordées pour les OD et les non-OD lors de l’évaluation initiale
Le degré de l’avantage supplémentaire accordé est lié aux preuves fournies. Il convient de noter que l’octroi d’une « prestation non ajoutée » ne s’applique pas aux DO dans l’évaluation des prestations, et que le degré par défaut de la prestation médicale ajoutée est « non quantifiable ». La plupart des évaluations initiales de la DO aboutissent à un bénéfice « non quantifiable » (figure 2). Pour les non-OD, la catégorie de prestations la plus attribuée était « aucune prestation ajoutée ». La répartition d’une prestation supplémentaire accordée dans les catégories « considérable » et « mineure » était plus ou moins comparable dans les évaluations initiales de DO et de non-OD.
Graphique 2. Avantages supplémentaires accordés lors des évaluations initiales

Graphique 2. Avantages supplémentaires accordés lors des évaluations initiales
Clé: Non. –nombre; OD – médicament orphelin.
L’évaluation des prestations entraîne une diminution des prix de remboursement (comme pour les OD et les non-OD)
Une analyse des niveaux de prix avant l’évaluation des avantages et après la négociation des prix a montré une réduction du prix de lancement par la procédure d’une évaluation initiale (OD : 18 % de rabais ; non-OD : 22 % de rabais). Une nouvelle cotisation a entraîné une nouvelle augmentation du remboursement, quoique dans une moindre mesure (remise de 8 % après réévaluation pour les OD et les non-OD). La différence de prix entre les OD et les non-OD est importante. En moyenne, les DO se sont avérés être environ 10,2 à 13,5 fois plus chers que les non-OD. Cette différence de coûts a persisté au lancement, après l’évaluation initiale des avantages et d’une réévaluation à l’autre (données non présentées).

Médicaments orphelins : Concilier les coûts élevés et les preuves solides
L’analyse a montré comment les OD, avec des prix généralement plus élevés et des preuves moins solides que les non-OD, sont évaluées dans le système de santé allemand. Bien que les DO ne constituent qu’une petite fraction de l’évaluation des prestations, leurs dépenses considérables, reflétées par les prix de remboursement élevés par rapport aux non-OD, soulignent leur impact significatif sur le système de santé. Le coût élevé des DO est influencé par de multiples facteurs, notamment la population restreinte de patients et les investissements substantiels en recherche et développement. En particulier, les coûts élevés des DO sont constamment discutés en termes de justification de leur prix par rapport aux preuves fournies, en particulier lorsque l’on considère le fardeau économique sur les systèmes de santé.
Les analyses ont révélé une comparabilité des données probantes présentées, les ECR constituant la base de données probantes pour la majorité des évaluations pour les personnes atteintes et les non-personnes atteintes de drogue. Comme le montrent les données, le statut de DO n’est pas nécessairement associé à un manque de qualité des preuves, qui a fait l’objet de débats. Cependant, la réduction du nombre d’études acceptées pour les DO dans la réévaluation souligne les difficultés de traiter le TCA. Comme on pouvait s’y attendre, la faible prévalence associée aux maladies rares rend difficile la réalisation d’ECR à grande échelle, ce qui met en évidence la corrélation entre la prévalence de la maladie et la qualité des données probantes. Cependant, il a été démontré que les sociétés pharmaceutiques soumettent des preuves de haute qualité, avec un nombre important d’ECR réalisés pour les OD, malgré les contraintes mentionnées ci-dessus.
En conclusion, les médicaments développés pour les maladies rares présentent des défis distincts pour les fabricants et les autorités de l’ETS. Bien que les « privilèges d’évaluation » améliorent la disponibilité des thérapies pour les maladies rares difficiles à traiter, la prudence est de mise lorsque des critères de médecine fondée sur des données probantes sont appliqués. Nos résultats démontrent que malgré les obstacles rencontrés par les OD, des preuves de haute qualité sous forme d’ECR sont fournies. Cependant, le système d’ETS reconnaît l’infaisabilité occasionnelle des ECR dans le domaine des maladies rares, tout en laissant une plus grande marge de manœuvre pour accepter les non-ECR dans les évaluations de la DO.
Bien que cet article ne se soit pas penché sur la justification des prix élevés des OD, leur corrélation d’efficacité ou la justification de l’avantage supplémentaire accordé, une évaluation plus approfondie est nécessaire pour analyser ces aspects clés et leur complexité au sein du système de santé allemand. L’interaction entre les coûts des OD, la force des données probantes qu’ils offrent et la valeur qu’ils apportent aux systèmes de soins de santé reste un sujet complexe et en constante évolution qui nécessite un examen et une analyse continus.
En conclusion, les médicaments développés pour les maladies rares présentent des défis distincts pour les fabricants et les autorités de l’ETS. Bien que les « privilèges d’évaluation » améliorent la disponibilité des thérapies pour les maladies rares difficiles à traiter, la prudence est de mise lorsque des critères de médecine fondée sur des données probantes sont appliqués. Nos résultats démontrent que malgré les obstacles rencontrés par les OD, des preuves de haute qualité sous forme d’ECR sont fournies. Cependant, le système d’ETS reconnaît l’infaisabilité occasionnelle des ECR dans le domaine des maladies rares, tout en laissant une plus grande marge de manœuvre pour accepter les non-ECR dans les évaluations de la DO.
Bien que cet article ne se soit pas penché sur la justification des prix élevés des OD, leur corrélation d’efficacité ou la justification de l’avantage supplémentaire accordé, une évaluation plus approfondie est nécessaire pour analyser ces aspects clés et leur complexité au sein du système de santé allemand. L’interaction entre les coûts des OD, la force des données probantes qu’ils offrent et la valeur qu’ils apportent aux systèmes de soins de santé reste un sujet complexe et en constante évolution qui nécessite un examen et une analyse continus.

Sources
-
Bundesministerium für Gesundheit (BMG). Seltene Erkrankungen. 2024. Consulté le 4 septembre 2024. https://www.bundesgesundheitsministerium.de/themen/praevention/gesundheitsgefahren/seltene-erkrankungen.html
-
Bundesministerium der Justiz (BMJ). Sozialgesetzbuch (SGB) Fünftes Buch (V). Consulté le 4 septembre 2024. https://www.gesetze-im-internet.de/sgb_5/__35a.html
-
Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen. Preis- und Kostenentwicklung von Orphan Drugs ; Arbeitspapier [online]. 2024. Consulté le 19 septembre 2024. https://dx.doi.org/10.60584/GA22-01


