Artikkel
Avsløring av bevisgapet: Sjeldne legemidler vs. ikke-sjeldne legemidler i tyske metodevurderinger
-
Kristina Dittrich, PhD
-
Nadine Klusmeier, PhD
-
Mareike Kubinski, PhD
-

Jan-Frederik Löpmeier-Röh, MSc
-

Susanne Kossow, PhD
-
Michael Reinartz, PhD
-

Werner Kulp, PhD
Legemidler for sjeldne sykdommer blir ofte forhåndsdømt til å mangle bevis fordi de har en garantert tilleggsfordel ved lov. Men er denne fordommen berettiget? Denne artikkelen tar deg med på en opplysende utforskning av medisinsk teknologivurdering i Tyskland for å belyse virkeligheten bak disse dvelende antakelsene.

Sjeldne sykdommer og deres behandlinger i tysk metodevurdering
Sjeldne sykdommer, som per definisjon rammer bare 1 av 2,000 mennesker i EU, mangler ofte effektive behandlingsalternativer. Sjeldne legemidler fyller dette gapet og får derfor spesielle insentiver for utvikling og kommersialisering. For markedsadgang i Tyskland får OD-er en unik behandling i medisinsk teknologivurdering (HTA).
Den tyske metodevurderingen for legemidler er regulert av «Arzneimittelmarktneuordnungsgesetz» (lov om markedsomorganisering av legemidler, AMNOG). AMNOG har som mål å balansere refusjon for legemiddelprodusenter og utgiftsbyrden for det nasjonale helsevesenet. Legemidler er forpliktet til å gjennomgå en nyttevurdering sammenlignet med en eksisterende standardbehandling (såkalt passende komparativ terapi [ACT]). En nyttevurdering er obligatorisk ved lansering av et nylig godkjent legemiddel med et nytt virkestoff i Tyskland eller ved nylig godkjente indikasjoner for et allerede vurdert legemiddel. Basert på den kliniske dokumentasjonen vurderer Federal Joint Committee (G-BA) den ekstra medisinske fordelen ved å bruke seks kategorier ('større', 'betydelig', 'mindre', 'ikke-kvantifiserbare', 'ingen ekstra fordel' eller 'mindre').
For OD-er gis en ekstra medisinsk fordel ved lov, og omfangsvurderingen er basert på sentral evidens uten forpliktelse til å sammenligne med en ACT. Men hvis salget når 30 millioner euro eller indikasjonen mister foreldreløsstatus, blir revurdering under standard evalueringsprosess obligatorisk. Ikke-sjeldne legemidler (ikke-OD), som er forpliktet til den regelmessige ytelsesvurderingen, kan også bli gjenstand for en revurdering, enten obligatorisk ved midlertidige beslutninger (f.eks. tilleggsdata etterspørres av G-BA) eller på forespørsel fra legemiddelprodusenten.
Nyttevurderingen ligger til grunn for prisforhandlingene for refusjon mellom legemiddelprodusenten og Landsforeningen for lovpålagte helseforsikringskasser (GKV-SV). Bare legemidler med ekstra medisinsk fordel kan oppnå en høyere pris enn kostnadene for ACT i prisforhandlingene. I løpet av de første seks månedene etter lansering står produsenten fritt til å definere lanseringsprisen, mens den forhandlede prisen brukes fra den syvende måneden. Refusjonsprisen for et legemiddel kan endres ytterligere med tiden når revurderinger og nye indikasjoner for det aktuelle legemidlet går gjennom HTA og påfølgende prisforhandlinger.
Denne artikkelen utforsker resultatene av en grundig analyse av tyske HTA-er som fokuserer på sammenligning av evidenskriterier og prising mellom OD-er og ikke-OD-er (både for innledende vurdering og revurdering) og tar sikte på å kaste lys over om OD-er mangler dokumentasjon i HTA sammenlignet med ikke-OD-er.
En omfattende gjennomgang analyserte nyttevurderinger for OD og ikke-OD fra 2011 til mai 2024 fra en database med G-BA-data. Denne analysen inkluderte 959 nyttevurderinger. Av disse var 810 innledende vurderinger av nylig godkjente legemidler eller nylig godkjente indikasjoner for et legemiddel, mens 149 var revurderinger, og noen produkter ble revurdert mer enn én gang for en respektive indikasjon. Analysen av 810 innledende nyttevurderinger avslørte at 27 % av vurderingene evaluerte OD, mens 73 % var ikke-OD. Totalt ble det utført en revurdering for 15 % av alle vurderinger (7 % OD og 8 % ikke-OD).
Eventuelle revurderinger ble matchet med den respektive innledende nyttevurderingen, hvis aktuelt, for å tilpasse relaterte vurderinger for samme legemiddel. Data ble hentet ut for OD-er og ikke-OD-er med fokus på parametere som evidensvurdert (studietyper, antall studier og størrelse på studier), nivå av tilkjent merfordel og pris. Evalueringene fokuserte på tilfeller med både innledende vurdering (60 OD og 64 ikke-OD) og revurdering (54 OD og 70 ikke-OD, noe som gjenspeiler at noen legemidler mistet OD-status fra et prevalensperspektiv og ble revurdert under standard prosedyre som ikke-OD) for en gitt indikasjon for å vurdere om dokumentasjon, nyttenivå og pris endret seg mellom innledende vurdering og revurdering, og å sammenligne mellom OD og ikke-OD.
For OD-er gis en ekstra medisinsk fordel ved lov, og omfangsvurderingen er basert på sentral evidens uten forpliktelse til å sammenligne med en ACT. Men hvis salget når 30 millioner euro eller indikasjonen mister foreldreløsstatus, blir revurdering under standard evalueringsprosess obligatorisk. Ikke-sjeldne legemidler (ikke-OD), som er forpliktet til den regelmessige ytelsesvurderingen, kan også bli gjenstand for en revurdering, enten obligatorisk ved midlertidige beslutninger (f.eks. tilleggsdata etterspørres av G-BA) eller på forespørsel fra legemiddelprodusenten.
Nyttevurderingen ligger til grunn for prisforhandlingene for refusjon mellom legemiddelprodusenten og Landsforeningen for lovpålagte helseforsikringskasser (GKV-SV). Bare legemidler med ekstra medisinsk fordel kan oppnå en høyere pris enn kostnadene for ACT i prisforhandlingene. I løpet av de første seks månedene etter lansering står produsenten fritt til å definere lanseringsprisen, mens den forhandlede prisen brukes fra den syvende måneden. Refusjonsprisen for et legemiddel kan endres ytterligere med tiden når revurderinger og nye indikasjoner for det aktuelle legemidlet går gjennom HTA og påfølgende prisforhandlinger.
Denne artikkelen utforsker resultatene av en grundig analyse av tyske HTA-er som fokuserer på sammenligning av evidenskriterier og prising mellom OD-er og ikke-OD-er (både for innledende vurdering og revurdering) og tar sikte på å kaste lys over om OD-er mangler dokumentasjon i HTA sammenlignet med ikke-OD-er.
En omfattende gjennomgang analyserte nyttevurderinger for OD og ikke-OD fra 2011 til mai 2024 fra en database med G-BA-data. Denne analysen inkluderte 959 nyttevurderinger. Av disse var 810 innledende vurderinger av nylig godkjente legemidler eller nylig godkjente indikasjoner for et legemiddel, mens 149 var revurderinger, og noen produkter ble revurdert mer enn én gang for en respektive indikasjon. Analysen av 810 innledende nyttevurderinger avslørte at 27 % av vurderingene evaluerte OD, mens 73 % var ikke-OD. Totalt ble det utført en revurdering for 15 % av alle vurderinger (7 % OD og 8 % ikke-OD).
Eventuelle revurderinger ble matchet med den respektive innledende nyttevurderingen, hvis aktuelt, for å tilpasse relaterte vurderinger for samme legemiddel. Data ble hentet ut for OD-er og ikke-OD-er med fokus på parametere som evidensvurdert (studietyper, antall studier og størrelse på studier), nivå av tilkjent merfordel og pris. Evalueringene fokuserte på tilfeller med både innledende vurdering (60 OD og 64 ikke-OD) og revurdering (54 OD og 70 ikke-OD, noe som gjenspeiler at noen legemidler mistet OD-status fra et prevalensperspektiv og ble revurdert under standard prosedyre som ikke-OD) for en gitt indikasjon for å vurdere om dokumentasjon, nyttenivå og pris endret seg mellom innledende vurdering og revurdering, og å sammenligne mellom OD og ikke-OD.

Sammenlignbarhet av presentert dokumentasjon for OD og ikke-OD
For OD-er gis det automatisk en ekstra fordel ved lov. Derfor, hvilke kliniske bevis presenteres for OD-er i nyttevurderinger, og hvordan skiller det seg fra ikke-OD-vurderinger? Som et første steg i standard nyttevurderinger vurderer G-BA kvaliteten på de innsendte studiene basert på kunnskapsnivået for å gi et tilstrekkelig pålitelig kunnskapsnivå. Basert på denne vurderingen er studien enten akseptert eller ikke vurdert i vurderingen. Videre må studier gitt for en standard nyttevurdering inkludere en sammenligning av legemidlet med ACT, definert av G-BA. Mens OD-er drar nytte av en mindre streng utførelse av beviskriterier for innledende vurderinger, blir de som overskrider salgsgrensen revurdert under standardprosedyren selv om de beholder foreldreløs status fra et prevalensperspektiv.
Evalueringene viste at randomiserte kontrollerte studier (RCT), gullstandarden for evidens, var den dominerende studietypen for både OD og ikke-OD (figur 1). For ikke-OD-er ble flere RCTer akseptert av G-BA i revurdering (51/70 tilfeller, 73 %) enn i innledende vurdering (44/64 tilfeller, 69 %), mens det var omvendt for OD-er (45/60 innledende vurderinger, 75 %; 35/54 revurderinger, 65 %).
Opprinnelig ble de kliniske studiene sendt inn av produsenten akseptert av G-BA i alle OD-vurderinger; flertallet av studiene (45/60 tilfeller, 75 %) var RCT, og 25 % (15/60 tilfeller, 25 %) var ikke-RCTer (f.eks. enarmsstudier, små studiestørrelser, bruk av surrogatutfall). Når OD-er ble revurdert i en standard fordelsvurdering, etter å ha overskredet salgsgrensen, sank andelen aksepterte RCT-er og ikke-RCT-er til henholdsvis 65 % (35/54 revurderinger) og 13 % (7/54 revurderinger). I 12 av OD-revurderingene (22 %) ble de respektive studiene ikke akseptert av G-BA, noe som gjenspeiler de strengere evidenskriteriene som ble brukt under standard vurderingsprosess. Å ikke oppfylle ACT kan være hovedårsaken til dette skiftet.
I motsetning til OD-er ble svært få ikke-RCT-er akseptert i ikke-OD-nyttevurderinger (2/64 innledende vurderinger; 1/70 revurderinger). Dermed ser det ut til at ikke-RCT-er er mer sannsynlig å bli akseptert for OD-er enn for ikke-OD-er. Videre ble produsentens bevis akseptert i alle OD innledende vurderinger, mens de presenterte studiene ble avvist i 18 vurderinger og revurderinger av ikke-OD. Dette skyldes det faktum at i OD-vurderinger er pivotale studier alltid akseptert ved lov.
Figur 1. Fordeling av studietype blant OD og ikke-OD i initial vurdering (IA) og revurdering (RA)
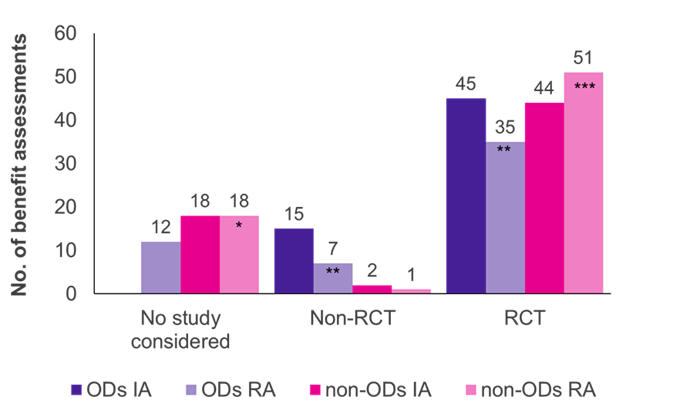
Nøkkel: IA – innledende vurdering; Nei. –nummer; OD – foreldreløst stoff; RA – revurdering; RCT – randomisert kontrollstudie.
Videre, og ikke overraskende, var det en positiv korrelasjon mellom økt størrelse på studiepopulasjoner og tilgjengeligheten av en RCT (data ikke vist).
Opprinnelig ble de kliniske studiene sendt inn av produsenten akseptert av G-BA i alle OD-vurderinger; flertallet av studiene (45/60 tilfeller, 75 %) var RCT, og 25 % (15/60 tilfeller, 25 %) var ikke-RCTer (f.eks. enarmsstudier, små studiestørrelser, bruk av surrogatutfall). Når OD-er ble revurdert i en standard fordelsvurdering, etter å ha overskredet salgsgrensen, sank andelen aksepterte RCT-er og ikke-RCT-er til henholdsvis 65 % (35/54 revurderinger) og 13 % (7/54 revurderinger). I 12 av OD-revurderingene (22 %) ble de respektive studiene ikke akseptert av G-BA, noe som gjenspeiler de strengere evidenskriteriene som ble brukt under standard vurderingsprosess. Å ikke oppfylle ACT kan være hovedårsaken til dette skiftet.
I motsetning til OD-er ble svært få ikke-RCT-er akseptert i ikke-OD-nyttevurderinger (2/64 innledende vurderinger; 1/70 revurderinger). Dermed ser det ut til at ikke-RCT-er er mer sannsynlig å bli akseptert for OD-er enn for ikke-OD-er. Videre ble produsentens bevis akseptert i alle OD innledende vurderinger, mens de presenterte studiene ble avvist i 18 vurderinger og revurderinger av ikke-OD. Dette skyldes det faktum at i OD-vurderinger er pivotale studier alltid akseptert ved lov.
Figur 1. Fordeling av studietype blant OD og ikke-OD i initial vurdering (IA) og revurdering (RA)
Nøkkel: IA – innledende vurdering; Nei. –nummer; OD – foreldreløst stoff; RA – revurdering; RCT – randomisert kontrollstudie.
Merk: Seks OD-er, opprinnelig vurdert som OD-er, ble revurdert som ikke-OD-er på grunn av tap av OD-status (*lagt til fire tidligere OD-er; **redusert med tre tidligere OD-er; ***lagt til to tidligere OD-er).
Videre, og ikke overraskende, var det en positiv korrelasjon mellom økt størrelse på studiepopulasjoner og tilgjengeligheten av en RCT (data ikke vist).
Fordeling av innvilgede merytelser for OD og ikke-OD i første vurdering
Graden av innvilget merfordel er knyttet til dokumentasjonen. Det er verdt å merke seg at det å gi "ingen ekstra fordel" ikke er aktuelt for OD-er i ytelsesvurderingen, og standardgraden av ekstra medisinsk fordel er "ikke-kvantifiserbar". De fleste OD-innledende vurderinger resulterer i en "ikke-kvantifiserbar" fordel (figur 2). For ikke-OD-er var den mest tildelte fordelskategorien 'ingen ekstra fordel'. Fordelingen av en innvilget tilleggsfordel i kategoriene «betydelig» og «mindre» var mer eller mindre sammenlignbar i OD- og ikke-OD-innledende vurderinger.
Figur 2. Innvilget ekstra fordel i innledende vurderinger

Figur 2. Innvilget ekstra fordel i innledende vurderinger
Nøkkel: Nei. –nummer; OD – orphan drug.
Ytelsesvurderinger resulterer i en reduksjon i refusjonsprisene (tilsvarende OD og ikke-OD)
En analyse av prisnivåene før fordelsvurdering og etter prisforhandling viste en reduksjon av lanseringsprisen gjennom prosedyren med en innledende vurdering (OD: 18 % rabatt; ikke-OD: 22 % rabatt). En revurdering førte til en ytterligere økning i rabatten, men i mindre grad (8 % rabatt etter revurdering for både OD og ikke-OD). Prisforskjellen mellom OD-er og ikke-OD-er er stor. I gjennomsnitt ble OD-er funnet å være omtrent 10,2 til 13,5 ganger dyrere enn ikke-OD-er. Denne kostnadsforskjellen vedvarte ved lansering, etter innledende vurdering av nytte og på tvers av revurderinger (data vises ikke).

Sjeldne stoffer: Balansere høye kostnader med sterke bevis
Analysen viste hvordan OD, med typisk høyere priser og mindre robust dokumentasjon enn ikke-OD, vurderes i det tyske helsevesenet. Selv om OD-er utgjør en liten brøkdel av ytelsesvurderingene, understreker deres betydelige utgifter, reflektert av de høye refusjonsprisene sammenlignet med ikke-OD-er, deres betydelige innvirkning på helsevesenet. De forhøyede kostnadene ved OD-er påvirkes av flere faktorer, spesielt den begrensede pasientpopulasjonen og de betydelige forsknings- og utviklingsinvesteringene. Spesielt blir de høye kostnadene ved OD-er gjentatte ganger diskutert med tanke på om prisen deres er berettiget i forhold til de fremlagte bevisene, spesielt når man vurderer den økonomiske byrden på helsevesenet.
Analysene indikerte sammenlignbarhet i den presenterte dokumentasjonen, med RCT som evidensgrunnlag for flertallet av vurderingene for både OD og ikke-OD. Som vist av dataene, er OD-statusen ikke nødvendigvis assosiert med mangel på kvalitet på bevis, som har vært gjenstand for debatter. Reduksjonen i aksepterte studier for OD-er i revurderingen understreker imidlertid vanskelighetene med å adressere ACT. Som forventet fører den lille prevalensen assosiert med sjeldne sykdommer til utfordringer med å gjennomføre omfattende RCT-er, noe som fremhever sammenhengen mellom sykdomsprevalens og kvalitet på dokumentasjon. Det ble imidlertid vist at farmasøytiske selskaper sender inn bevis av høy kvalitet, med et betydelig antall RCTer utført for OD, til tross for begrensningene nevnt ovenfor.
Konklusjonen er at legemidler utviklet for sjeldne sykdommer utgjør klare utfordringer for produsenter og HTA-myndigheter. Mens "vurderingsprivilegier" forbedrer tilgjengeligheten av behandlinger for sjeldne tilstander som er vanskelige å behandle, er det nødvendig med forsiktighet når evidensbaserte medisinkriterier brukes. Våre funn viser at til tross for hindringene som OD står overfor, er det gitt bevis av høy kvalitet i form av RCT. HTA-systemet anerkjenner imidlertid at RCT av og til er ugjennomførbart innen sjeldne sykdommer, samtidig som det gir mer spillerom til å akseptere ikke-RCT i OD-vurderinger.
Selv om denne artikkelen ikke fordypet seg i begrunnelsen for ODs høye priser, deres effektkorrelasjon eller begrunnelsen bak den innvilgede tilleggsfordelen, er ytterligere evaluering berettiget for å analysere disse nøkkelaspektene og deres kompleksitet i det tyske helsevesenet. Samspillet mellom kostnadene ved OD, styrken på bevisene de tilbyr, og verdien de tilfører helsevesenet, er fortsatt et komplekst og utviklende tema som krever kontinuerlig gransking og analyse.
Konklusjonen er at legemidler utviklet for sjeldne sykdommer utgjør klare utfordringer for produsenter og HTA-myndigheter. Mens "vurderingsprivilegier" forbedrer tilgjengeligheten av behandlinger for sjeldne tilstander som er vanskelige å behandle, er det nødvendig med forsiktighet når evidensbaserte medisinkriterier brukes. Våre funn viser at til tross for hindringene som OD står overfor, er det gitt bevis av høy kvalitet i form av RCT. HTA-systemet anerkjenner imidlertid at RCT av og til er ugjennomførbart innen sjeldne sykdommer, samtidig som det gir mer spillerom til å akseptere ikke-RCT i OD-vurderinger.
Selv om denne artikkelen ikke fordypet seg i begrunnelsen for ODs høye priser, deres effektkorrelasjon eller begrunnelsen bak den innvilgede tilleggsfordelen, er ytterligere evaluering berettiget for å analysere disse nøkkelaspektene og deres kompleksitet i det tyske helsevesenet. Samspillet mellom kostnadene ved OD, styrken på bevisene de tilbyr, og verdien de tilfører helsevesenet, er fortsatt et komplekst og utviklende tema som krever kontinuerlig gransking og analyse.

Kilder
-
Bundesministerium für Gesundheit (BMG). Seltene Erkrankungen. 2024. Åpnet 4. september 2024. https://www.bundesgesundheitsministerium.de/themen/praevention/gesundheitsgefahren/seltene-erkrankungen.html
-
Bundesministerium der Justiz (BMJ). Sozialgesetzbuch (SGB) fünftes Buch (V). Åpnet 4. september 2024. https://www.gesetze-im-internet.de/sgb_5/__35a.html
-
Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen. Preis- und Kostenentwicklung von Orphan Drugs; Arbeitspapier [online]. 2024. Åpnet 19 september 2024. https://dx.doi.org/10.60584/GA22-01


